The right answers to frequently asked questions
Find the answers to all our products and services by clicking the links below.
General Questions regarding Sample Submission, Ordering & Sample Shipment & Results can be found here:
What kind of quality control do you perform?
All ready-to-load library pools are subject to a quality and quantity control to accurately measure the insert size and DNA concentration to determine appropriate sequencing dilution and achieve optimal cluster densities. .
What should I do if my sample fails the entry QC?
If the amount, concentration or quality of the ready-to-sequence library does not meet the requirements for further sample processing, we will contact you to discuss how to go on with the project. Whenever possible, Eurofins Genomics will make recommendations for optimisation of sample quality.
Do you guarantee a certain number of reads?
The actual achieved sequencing output (read length, read quality and number of reads) is directly correlated with the quality of the sequencing library used. Eurofins Genomics cannot give any guarantees for sequencing data resulting from ready-to-load libraries prepared by the customer. If unsatisfactory sequencing results are due to technical problems with the sequencing kits or machines, the sequencing will be repeated at no additional cost to the customer.
Is the sorting of the reads included?
Raw data sorting according to Illumina Index read is free of charge. Used index type (single-indexed or (unique) dual-indexed) and corresponding index sequences have to be provided prior to project start in the Sample Submission Form.
Can Eurofins perform de-multiplexing of “in-line” barcodes in sequencing reads?
Yes, we offer de-multiplexing of in-line barcodes in sequencing reads (not index reads). We highly recommend to avoid the “in-line” barcoding strategy and use the Illumina index system instead (i.e. the barcodes are read in a separate read and do not interfere with cluster registration). It is important to ensure that the base composition of the indices is balanced to optimise the ability of the image analysis software to distinguish signals.
Please contact us for further details. Additional charges apply
What type of ready-to-load library pool can I submit?
You can submit any library pool compatible with Illumina® sequencing, including those prepared from genomic DNA for whole genome sequencing, RNA for transcriptome analysis, or amplicon libraries. Please note, pooling of different libraries into a final ready-to-load pool must be performed by our customers, this service is not offered by Eurofins Genomics.
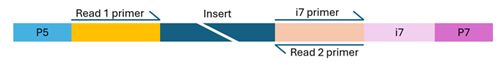
Illumina-compatible libraries must include four key components in their adapter sequences—located at the ends of each fragment—for successful paired-end sequencing:
- Flow cell binding sites (P5 and P7): These enable the DNA fragments to attach and cluster on the flow cell.
- Sequencing primer binding sites (Read 1 and Read 2): These sites initiate sequencing from both ends of the DNA fragment.
- Index sequences (i5 and i7): Also known as barcodes, these allow for sample multiplexing. Libraries may be single-indexed (one index) or dual-indexed (two indexes). The usual length of barcodes for NovaSeq is in the range of 8 to 12 bp, while for MiSeq we recommend to use at maximum 10 bp index sequences.
- Index primer binding site(s): These are essential for reading the index sequences during sequencing.
Single-indexed library
Dual-indexed library
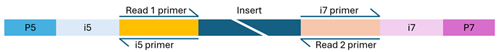
The structure of the adapter sequences varies depending on whether the library uses single or dual indexing. Missing, misconfigured, or heavily modified components can cause sequencing to fail. If you're unsure about your library’s configuration, feel free to contact us for guidance.
Can you spike PhiX into my sequencing run?
Illumina cluster detection algorithms are optimized around a balanced representation of A, T, G, and C nucleotides. Any divergence from equal base distribution will negatively influence the amount and quality of sequencing data produced. To increase the library nucleotide balance a spike-in of 20% PhiX (MiSeq) will be used for samples with low diversity or unbalanced base composition (e.g., amplicons, bisulfite converted samples). The extent to which the negative impact of the unbalanced base composition will be reduced by spiking the PhiX control depends on individual sample and sequence characteristics. For more information please refer to the Illumina website.
Can you perform data analysis for my ready-to-load project?
Yes, we can do optional data analysis. Please indicate this on the quote request form.